









Sisällys
Bournevillen tuberoosiskleroosi
Mikä se on ?
Bournevillen tuberoosiskleroosi on monimutkainen geneettinen sairaus, jolle on tunnusomaista hyvänlaatuisen (ei-syöpäkasvaimen) kehittyminen kehon eri osissa. Nämä kasvaimet voivat sitten sijaita iholla, aivoissa, munuaisissa ja muissa elimissä ja kudoksissa. Tämä patologia voi myös aiheuttaa vakavia ongelmia yksilön kehityksessä. Taudin kliiniset ilmenemismuodot ja vakavuus vaihtelevat kuitenkin potilaittain.
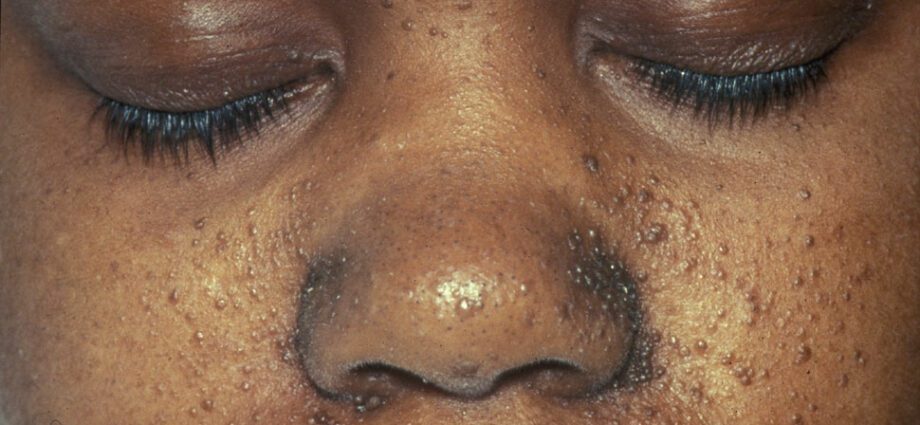
Tähän liittyvät ihon poikkeavuudet ovat yleensä samanlaisia kuin ihon täplät tai alueet, joilla iho on vaaleampi kuin muualla kehossa. Kasvainten kehittymistä kasvoissa kutsutaan angiofibromaksi.
Aivovaurion yhteydessä kliiniset oireet ovat epileptisiä kohtauksia, käyttäytymisongelmia (yliaktiivisuus, aggressiivisuus, kehitysvamma, oppimisvaikeudet jne.). Joillakin sairastuneilla lapsilla on jopa jonkinlainen autismi, kehityshäiriöt, jotka vaikuttavat sosiaaliseen vuorovaikutukseen ja viestintään. Hyvänlaatuiset aivokasvaimet voivat myös aiheuttaa komplikaatioita, jotka voivat olla kohtalokkaita potilaalle.
Kasvainten kehittyminen munuaisissa on yleistä tuberoosiskleroosipotilailla. Tämä voi aiheuttaa vakavia komplikaatioita munuaisten toiminnassa. Lisäksi kasvaimia voi kehittyä sydämeen, keuhkoihin ja verkkokalvoon. (2)
Se on harvinainen sairaus, jonka esiintyvyys (tapausten määrä tietyssä populaatiossa tiettynä ajankohtana) on 1/8 000 1 ihmistä. (15)
oireet
Bournevillen tuberoosiskleroosiin liittyvät kliiniset oireet vaihtelevat elinten mukaan. Lisäksi sairauteen liittyvät oireet vaihtelevat suuresti yksilöstä toiseen. Oireet vaihtelevat lievästä vaikeaan.
Tämän taudin yleisimmin tunnistettuja oireita ovat epileptiset kohtaukset, kognitiiviset ja käyttäytymishäiriöt, ihon poikkeavuudet jne. Useimmin vaikuttavat elimet ovat: aivot, sydän, munuaiset, keuhkot ja iho.
Pahanlaatuisten (syöpä) kasvainten kehittyminen on mahdollista tässä taudissa, mutta ne ovat harvinaisia ja vaikuttavat pääasiassa munuaisiin.
Aivojen taudin kliiniset oireet ovat peräisin eri tasojen hyökkäyksistä:
- aivokuoren vaurioita;
- ependymaaliset kyhmyt (SEN);
- jättimäiset ependymaaliset astrosytoomat.
Ne johtavat: kehitysvammaisuuden kehittymiseen, oppimisvaikeuksiin, käyttäytymishäiriöihin, aggressiivisuuteen, tarkkaavaisuushäiriöihin, yliaktiivisuuteen, pakko-oireisiin häiriöihin jne.
Munuaisvaurioille on ominaista kystien tai angiomyolipoomien kehittyminen. Nämä voivat johtaa munuaiskipuun ja jopa munuaisten vajaatoimintaan. Jos runsasta verenvuotoa on havaittavissa, se voi johtua vaikeasta anemiasta tai korkeasta verenpaineesta. Myös muita vakavampia mutta harvinaisia seurauksia voi olla näkyvissä, erityisesti karsinoomien (epiteelin ainesosien kasvain) kehittyminen.
Silmävaurio voi olla samanlainen kuin verkkokalvon näkyvät täplät, aiheuttaen näköhäiriöitä tai jopa sokeutta.
Ihon poikkeavuuksia on lukuisia:
- hypomelaniset makulat: jotka aiheuttavat kevyitä täpliä iholla kaikkialla kehossa melaniinin puutteen seurauksena, joka antaa iholle väriä;
- punaisten pisteiden esiintyminen kasvoilla;
- värjäytyneet laastarit otsassa;
- muut ihosairaudet, jotka ovat riippuvaisia yksilöstä toiseen.
Keuhkovaurioita esiintyy 1/3: lla potilaista, joilla on lievä naisvaltaisuus. Siihen liittyvät oireet ovat silloin enemmän tai vähemmän vakavia hengitysvaikeuksia.
Taudin alkuperä
Taudin alkuperä on perinnöllinen ja perinnöllinen.
Lähetykseen liittyy mutaatioita TSC1- ja TSC2 -geeneissä. Nämä kiinnostavat geenit tulevat pelaamaan proteiinien muodostamisessa: hamartiini ja tuberiini. Nämä kaksi proteiinia mahdollistavat interaktiivisen pelin kautta solujen lisääntymisen säätelyn.
Potilailla, joilla on sairaus, syntyy vähintään yksi mutatoitu kopio näistä geeneistä kussakin solussa. Nämä mutaatiot rajoittavat hamartiinin tai tubertiinin muodostumista.
Kun geenin kaksi kopiota mutatoidaan, ne estävät täysin näiden kahden proteiinin tuotannon. Tämä proteiinipuutos ei siis enää salli kehon säätää tiettyjen solujen kasvua ja johtaa tässä mielessä kasvainsolujen kehittymiseen eri kudoksissa ja / tai elimissä.
Riskitekijät
Riskitekijät tällaisen patologian kehittymiselle ovat geneettisiä.
Itse asiassa taudin siirto on tehokasta autosomaalisen hallitsevan tilan kautta. Joko kiinnostuksen kohteena oleva mutatoitunut geeni sijaitsee ei-seksuaalisessa kromosomissa. Lisäksi vain yhden esiintyminen kahdesta mutatoidun geenin kopiosta riittää taudin kehittymiseen.
Tässä mielessä henkilöllä, jolla on yksi näistä kahdesta vanhemmista, jotka kärsivät sairaudesta, on 50% riski sairastua itse sairas fenotyyppiin.
Ehkäisy ja hoito
Taudin diagnoosi on ensinnäkin erilainen. Se perustuu epätyypillisiin fyysisiin kriteereihin. Useimmissa tapauksissa taudin ensimmäiset tunnusomaiset merkit ovat: toistuvien epileptisten kohtausten esiintyminen ja kohteen kehityksen viivästyminen. Muissa tapauksissa nämä ensimmäiset merkit johtavat ihottumiin tai sydänkasvaimen tunnistamiseen.
Tämän ensimmäisen diagnoosin jälkeen lisätutkimukset ovat välttämättömiä diagnoosin vahvistamiseksi. Nämä sisältävät:
- aivojen skannaus;
- aivojen magneettikuvaus (MRI);
- sydämen, maksan ja munuaisten ultraääni.
Diagnoosi voi olla tehokas lapsen syntymän yhteydessä. Muussa tapauksessa on tärkeää, että se suoritetaan mahdollisimman nopeasti, jotta potilas voidaan hoitaa mahdollisimman pian.
Tällä hetkellä tautiin ei ole parannuskeinoa. Siksi niihin liittyvät hoidot ovat riippumattomia kunkin yksilön esittämistä oireista.
Yleensä epilepsialääkkeitä annetaan kouristusten rajoittamiseksi. Lisäksi määrätään myös lääkkeitä aivojen ja munuaisten kasvainsolujen hoitoon. Käyttäytymisongelmien yhteydessä lapsen erityiskohtelu on tarpeen.
Taudin hoito on yleensä pitkäaikaista. (1)