









Sisällys
Progeria tai Hutchinson-Gilfordin oireyhtymä
Progeria on harvinainen geneettinen sairaus, jolle on ominaista lapsen varhainen ikääntyminen.
Määritelmä progeria
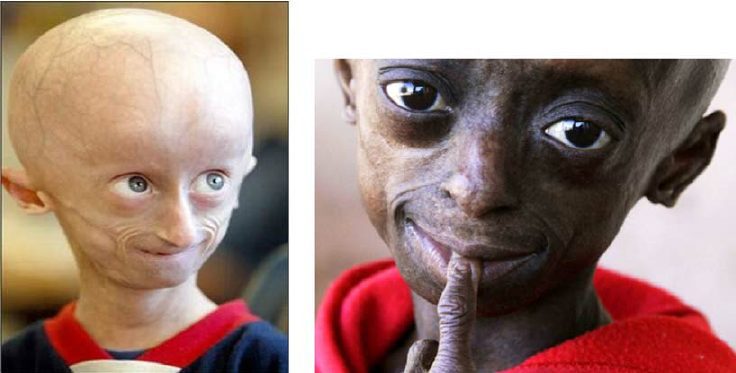
Progeria, joka tunnetaan myös nimellä Hutchinson-Gilfordin oireyhtymä, on harvinainen geneettinen sairaus. Sille on ominaista lisääntynyt kehon ikääntyminen. Lapset, joilla on tämä patologia, osoittavat varhaisia merkkejä vanhuudesta.
Syntymähetkellä lapsella ei ole poikkeavuuksia. Se näyttää "normaalilta" lapsuuteen asti. Vähitellen hänen morfologiansa ja kehonsa kehittyvät epänormaalisti: hän kasvaa normaalia hitaammin eikä lisää ikänsä lapsen painoa. Kasvoissa kehitys myös viivästyy. Hänellä on näkyvät silmät (helpotuksessa, voimakkaasti edistyneet), hyvin ohut ja koukussa oleva nenä, ohuet huulet, pieni leuka ja ulkonevat korvat. Progeria on myös syy merkittävään hiustenlähtöön (hiustenlähtöön), ihon ikääntymiseen, nivelvajeisiin tai jopa ihonalaisen rasvan menetykseen (ihonalainen rasva).
Lapsen kognitiiviseen ja älylliseen kehitykseen ei yleensä vaikuta. Se on olennaisesti puute ja vaikutuksia motoriseen kehitykseen, mikä vaikeuttaa istumista, seisomista tai jopa kävelyä.
Potilailla, joilla on Hutchinson-Gilfordin oireyhtymä, on myös joidenkin valtimoiden kapenemista, mikä johtaa ateroskleroosin kehittymiseen (valtimoiden tukos). Kuitenkin ateroskleroosi on hallitseva tekijä lisääntyneessä sydänkohtauksen tai jopa aivoverenkiertohäiriön (aivohalvauksen) riskissä.
Tämän taudin esiintyvyys (sairastuneiden määrä koko väestöstä) on 1/4 miljoonaa vastasyntynyttä maailmanlaajuisesti. Se on siis harvinainen sairaus.
Koska progeria on autosomaalinen hallitseva perinnöllinen sairaus, suurimmat riskit tällaisen sairauden kehittymiselle ovat ne, joiden vanhemmilla on myös progeria. Satunnaisten geneettisten mutaatioiden riski on myös mahdollinen. Progeria voi sitten vaikuttaa keneen tahansa, vaikka tauti ei olisi läsnä perhepiirissä.
Progerian syyt
Progeria on harvinainen ja geneettinen sairaus. Tämän patologian alkuperä johtuu LMNA -geenin mutaatioista. Tämä geeni on vastuussa proteiinin muodostumisesta: Lamine A. Jälkimmäisellä on tärkeä rooli solun ytimen muodostumisessa. Se on olennainen osa ydinkuoren muodostumista (solujen ydintä ympäröivä kalvo).
Tämän geenin geneettiset mutaatiot johtavat Lamine A: n epänormaaliin muodostumiseen. Tämä epänormaalisti muodostunut proteiini johtuu solun ytimen epävakaudesta ja organismin solujen varhaisesta kuolemasta.
Tutkijat kysyvät parhaillaan itseltään aihetta saadakseen lisätietoja tämän proteiinin osallistumisesta ihmiskehon kehitykseen.
Hutchinson-Gilfordin oireyhtymän geneettinen siirto tapahtuu autosomaalisen hallitsevan perinnön kautta. Joko vain yhden spesifisen geenin kahdesta kopiosta siirtyminen (joko äidiltä tai isältä) riittää taudin kehittymiseen lapsessa.
Lisäksi LMNA -geenin satunnaiset mutaatiot (jotka eivät johdu vanhempien geenien siirrosta) voivat olla myös tällaisen sairauden alkuperää.
Progerian oireet
Progerian yleisille oireille on ominaista:
- kehon varhainen ikääntyminen (lapsuudesta lähtien);
- vähemmän painonnousua kuin normaalisti;
- lapsen pienempi koko;
- kasvojen poikkeavuudet: ohut, koukussa oleva nenä, näkyvät silmät, pieni leuka, ulkonevat korvat jne.
- moottorin viivästyminen, mikä vaikeuttaa seisomista, istumista tai jopa kävelyä;
- valtimoiden kaventuminen, mikä on merkittävä ateroskleroosin riskitekijä.
Progerian riskitekijät
Koska progeria on harvinainen, geneettinen ja perinnöllinen autosomaalinen hallitseva sairaus, taudin esiintyminen toisessa vanhemmista on tärkein riskitekijä.
Mikä hoito progerialle?
Progeriaan liittyvät oireet ovat eteneviä ja voivat johtaa potilaan kuolemaan.
Sairauteen ei ole tällä hetkellä parannuskeinoa. Ainoa progerian hoito on oireiden hoito.
Tutkimus on tällöin valokeilassa, jotta löydettäisiin hoito, joka mahdollistaa tällaisen sairauden parantamisen.