









talassemia
Talassemiat ovat joukko perinnöllisiä verisairauksia, jotka vaikuttavat hemoglobiinin (hapen kuljettamisesta vastaavan proteiinin) tuotantoon. Ne vaihtelevat vakavuudeltaan: jotkut eivät aiheuta oireita, kun taas toiset ovat hengenvaarallisia. Luuytimensiirtoa harkitaan vakavimmissa tapauksissa.
Talassemia, mikä se on?
Määritelmä talassemia
Talassemialle on ominaista hemoglobiinin tuotannon häiriö. Muistutuksena, hemoglobiini on suuri punasoluissa (punasoluissa) oleva proteiini, jonka tehtävänä on varmistaa diksoygeenin kuljetus hengityselimistä muualle elimistöön.
Sanotaan, että talassemia on verisairaus. Punasolujen kuljetustoiminto on heikentynyt, millä voi olla vakavia seurauksia kehossa. Tässä vaiheessa on tärkeää huomata, että on olemassa useita talassemiatyyppejä, joilla ei ole samoja ominaisuuksia tai samaa vakavuusastetta. Joillakin ei ole oireita, kun taas toiset ovat hengenvaarallisia.
Talassemian syyt
Talassemiat ovat geneettisiä sairauksia. Ne johtuvat yhden tai useamman hemoglobiinin synteesiin osallistuvan geenin muutoksesta ja tarkemmin sanoen hemoglobiinin proteiiniketjujen tuotantoon osallistuvien geenien muutoksesta. Näitä on neljä: kaksi alfaketjua ja kaksi beetaketjua.
Jokainen näistä ketjuista voi vaikuttaa talassemiaan. Voimme myös erottaa:
- alfa-talassemiat, joille on tunnusomaista alfa-ketjun muutos;
- beeta-talassemiat, joille on ominaista beeta-ketjun muutos.
Alfa- ja beetatalassemioiden vakavuus riippuu muuttuneiden geenien määrästä. Mitä tärkeämpi se on, sitä suurempi vakavuusaste.
Talassemian diagnoosi
Talassemia diagnosoidaan verikokeella. Täydellinen verenkuva mahdollistaa punasolujen ulkonäön ja lukumäärän arvioinnin ja siten hemoglobiinin kokonaismäärän selvittämisen. Hemoglobiinin biokemialliset analyysit tekevät mahdolliseksi erottaa alfa-talassemiat beetatalassemioista. Lopuksi geneettisten analyysien avulla voidaan arvioida muuttuneiden geenien lukumäärä ja siten määrittää talassemian vakavuus.
Asianomaiset henkilöt
Talassemiat ovat perinnöllisiä geneettisiä sairauksia, eli ne siirtyvät vanhemmilta lapsilleen. Ne tavoittavat ihmiset pääasiassa Välimeren reunalta, Lähi-idästä, Aasiasta ja Saharan eteläpuolisesta Afrikasta.
Ranskassa alfa-talassemiaa esiintyy arviolta yhdellä ihmisellä 1:stä. Beetatalassemian ilmaantuvuuden arvioidaan olevan 350 syntymää per vuosi maailmanlaajuisesti.
Talassemian oireet
Talassemian oireet vaihtelevat suuresti tapauskohtaisesti ja riippuvat pääasiassa hemoglobiinin proteiiniketjujen tuotantoon osallistuvien geenien muutosasteesta. Talassemiat voivat olla vähäisissä muodoissaan oireettomia ja vakavammissa muodoissaan hengenvaarallisia.
Alla mainitut oireet koskevat vain talassemian keskimuotoisia ja suuria muotoja. Nämä ovat vain tärkeimmät oireet. Hyvin spesifisiä oireita voidaan joskus nähdä talassemian tyypistä riippuen.
Anemia
Tyypillinen talassemian merkki on anemia. Tämä on hemoglobiinin puute, joka voi johtaa erilaisiin oireisiin:
- väsymys
- hengenahdistus;
- kalpeus;
- epämukavuus;
- sydämentykytys.
Näiden oireiden voimakkuus vaihtelee talassemian vakavuudesta riippuen.
keltatauti
Talassemiaa sairastavilla ihmisillä voi olla keltaisuutta (keltaisuutta), joka näkyy iholla tai silmänvalkuaisissa.
sappikivet
Kivien muodostumista sappirakon sisällä voidaan myös nähdä. Laskelmat ovat kuin "pieniä kiviä".
splenomegaly
Splenomegalia on pernan laajentuma. Yksi tämän elimen tehtävistä on suodattaa verta ja suodattaa haitallisia aineita, mukaan lukien epänormaalit punasolut. Talassemiassa perna on voimakkaasti mobilisoitunut ja sen koko kasvaa vähitellen. Kipu voi tuntua.
Muut, harvinaisemmat oireet
Harvemmin vakavat talassemian muodot voivat johtaa muihin poikkeavuuksiin. Sen voi havaita esimerkiksi:
- hepatomegalia, eli maksan koon kasvu;
- luun epämuodostumat;
- viivästynyt lapsen kehitys;
- haavaumat.
Talassemian hoito on välttämätöntä näiden komplikaatioiden esiintymisen rajoittamiseksi.
Talassemian hoidot
Talassemian hoito riippuu monista parametreistä, mukaan lukien talassemian tyyppi, sen vakavuus ja asianomaisen henkilön tila. Pienet muodot eivät vaadi hoitoa, kun taas vaikeat muodot vaativat erittäin säännöllistä lääketieteellistä seurantaa.
Alla mainitut hoidot koskevat vain talassemian välimuotoja suuriin muotoihin
Anemian korjaus
Kun hemoglobiinin puute on liian suuri, tarvitaan säännöllisiä verensiirtoja. Niihin kuuluu luovuttajan veren tai punasolujen injektointi asianomaiselle henkilölle, jotta veren punasolujen määrä pysyy hyväksyttävänä.
B9-vitamiinilisä
Voidaan suositella päivittäisen B9-vitamiinilisän aloittamista, koska tämän vitamiinin tarve on lisääntynyt talassemiatapauksissa. B9-vitamiini osallistuu punasolujen tuotantoon.
splenektomiaan
Splenektomia on pernan kirurginen poisto. Tätä toimenpidettä voidaan harkita, kun anemia on erittäin tärkeä.
Raudan ylikuormituksen hoito
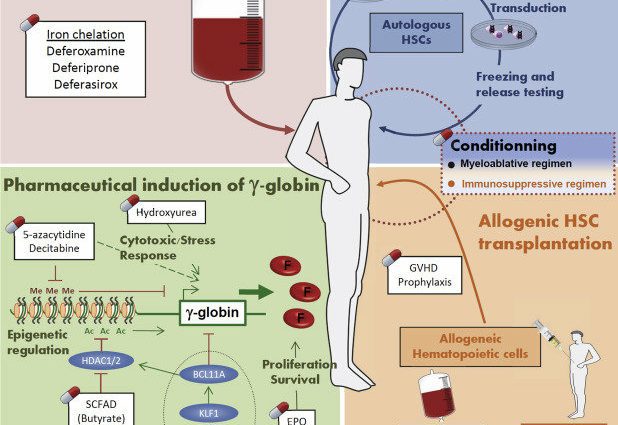
Talassemiaa sairastavien ihmisten kehossa on ylikuormitettu rautaa. Tämä kasaantuminen voi johtaa erilaisiin komplikaatioihin. Tästä syystä rautakelaattoreita tarjotaan poistamaan ylimääräistä rautaa.
Luuytimensiirto
Luuytimensiirto on ainoa hoito, joka voi parantaa talassemiaa pysyvästi. Tämä on raskas hoito, jota tarjotaan vain taudin vakavimmissa muodoissa.
Estä talassemia
Talassemia on perinnöllinen geneettinen sairaus. Ennalta ehkäisevää toimenpidettä ei ole.
Toisaalta geenitestien avulla voidaan havaita terveet kantajat (henkilöt, joilla on yksi tai useampi muuttunut geeni, mutta jotka eivät ole sairaita). Muutamille terveille kantajille tulee kertoa riskistä synnyttää talassemiaa sairastava lapsi. Joissakin tapauksissa geneetikko voi arvioida tämän riskin. Prenataalidiagnoosia voidaan myös harkita tietyissä olosuhteissa. Asiasta tulee keskustella lääkärisi kanssa.